기존 화합물 생성 AI 연구의 문제점 극복… 효과 증진한 새로운 약물 구조 자동 생성 기대
광주과학기술원(GIST, 총장 임기철) 전기전자컴퓨터공학부 남호정 교수 연구팀이 신규 약물 구조를 생성해주는 인공지능 모델을 개발했다.

생성형 인공지능 모델을 이용하는 신약 디자인 플랫폼으로, 약물의 효과를 최적화한 저분자 화합물 생성을 통해 신약 개발 시간을 획기적으로 단축하는 데 기여할 것으로 기대된다.
최근 다양한 인공지능 기반의 약물 디자인 플랫폼들이 등장하고 있는 가운데 대용량 화합물 데이터 기반 생성형 인공지능 모델을 훈련해 기존에 존재하지 않던 신규 구조의 약물을 디자인(De novo drug design) 하거나 선도물질을 최적화(Lead optimization)* 하는 연구들이 주목받고 있다.
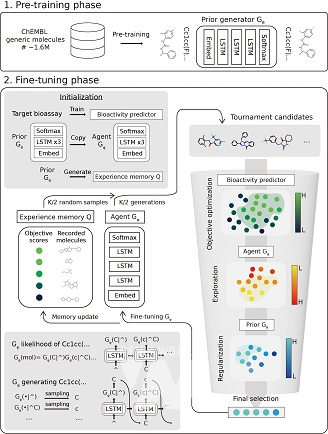
특히, 인공지능 모델 학습에 사용될 데이터가 부족한 상황에서도 활용이 가능한 전이 학습(Transfer learning)*을 도입하는 연구가 활발히 진행되고 있다. 이 방식은 먼저 화합물 구조를 언어적 문법으로 해석해 대량의 화합물 데이터를 사전학습한 후(Pre-trained model), 적은 수의 약물 활성 데이터를 이용하여 미세 조정(Fine-tuning)*을 진행한다.
* 선도물질 최적화(Lead optimization): 신약을 개발하기 위해 선도 물질(질병을 일으키는 물질을 제어할 수 있는 신약 후보)을 찾아내어 생물학적 활성이 뛰어난 물질로 개발하는 과정이다.
* 전이 학습(Transfer learning): 특정한 목적에 맞는 인공지능 모델을 학습하기 위해, 이전에 이미 다른 목적으로 정제된 데이터로 훈련을 마친 모델의 일부분을 가져와서 새로운 목적에 맞게 재학습 시키는 것을 의미한다.
* 미세 조정(Fine-tuning): 기존 데이터는 기존대로 분류하고 새로운 데이터로 다시 한 번 가중치를 세밀하게 조정하도록 학습하는 것으로 이 모델은 주어진 문제가 모델을 학습시킨 화합물과 매우 다른 경우에 더 효과적이며, 데이터 셋이 작을 때 이점이 있으며 빠르게 최적화할 수 있다.
기존의 전이 학습을 이용한 연구들은 미세 조정 단계에서 생성물의 구조적 다양성 부족해 모드 붕괴(Mode collapse)* 현상이 일어나 생성모델이 최적의 분포(Optimal distribution)를 학습하는데 어려움이 있다. 이는 결과적으로 다양한 신규 약물 구조를 생성하는 데 한계로 작용한다.
* 모드 붕괴(Mode collapse) 생성형 인공지능을 훈련할 때 발생 가능한 현상으로, 생성모델이 다양한 형태/구조의 생성물을 만들어내지 못하고, 반복적으로 유사한 생성물만 출력하는 현상을 의미한다.
전통적인 신약개발 과정에서 선도물질의 최적화 단계는 수개월부터 수년까지 걸릴 수 있는 긴 연구 과정을 필요로 한다. 그러나 이 단계에 인공지능을 도입하면 개발 시간이 몇 주에서 몇 개월로 크게 줄어들 수 있어 인공지능 기술의 활용이 크게 주목받고 있다.
특히 이번에 개발된 인공지능 모델은 특정 표적 단백질에 특화된 모델이 아닌 다양한 치료제 개발에 적용될 수 있도록 표적 단백질 정보를 쉽게 변경 가능하도록 설계되었다는 큰 장점이 있어 범용적인 약물 개발 시장에 적용될 수 있을 것으로 기대된다.
연구팀은 신규 약물 구조 디자인을 위한 최적의 생성 분포 학습 기술(LOGICS: Learning optimal generative distribution for designing de novo chemical structures)을 개발했다.
연구팀은 기존의 전이 학습 모델의 문제점을 해결하기 위해 미세조정 단계에서 경험 기억 메모리(Experience memory)와 토너먼트 선택(Tournament selection)을 이용하는 방식을 고안해 생성모델이 더욱 다양한 화합물 구조를 탐색할 수 있는 훈련 알고리즘을 제안했다.
이번 연구는 실제 약물 분자로부터의 화학적 거리를 측정하는 지표인 FCD*를 기준으로, 이전에 소개된 대표적인 신약 개발 플랫폼들(REINVENT, DrugEx 등)과 비교했을 때 우수한 성능(REINVENT의 32.7 FCD 대비 29.4 FCD로 향상된 성능 확인)을 보였고, 신규 화합물들이 실제 활성 약물과 더욱 유사한 속성을 가지고 있음을 검증했다.
* FCD(Fréchet ChemNet Distance): 실제 약물 분자로부터의 화학적 거리 지표, 값이 작을수록 기준으로 설정된 실제 약물들과의 유사성이 높은 분자를 생성할 확률이 높다.
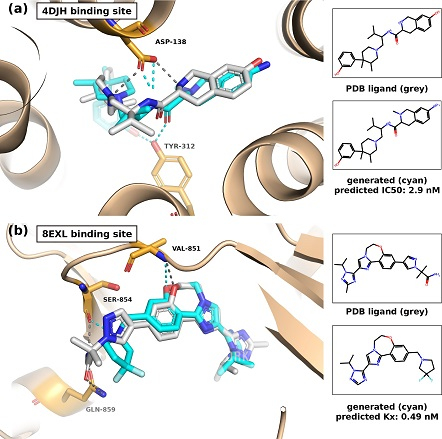
연구팀이 개발한 인공지능 모델이 제시한 분자 구조를 분석한 결과, 약물 후보로 예측된 다수의 생성물이 기존의 데이터에 있는 분자와는 유사성이 낮은 신규 구조임을 밝혔다.
또한 기존 화합물의 주요 골격은 유지하되, 세부 구조의 변화를 통해 표적 활성도 및 표적 단백질과의 3차원 구조적 결합을 향상하는 약물 최적화 방식도 적용 가능할 것으로 확인됐다.
남호정 교수는 “이번 연구 성과는 기존 전이학습 모델의 문제점을 해결해 안정적인 학습을 유도할 수 있으며, 데이터가 한정된 상황에서도 고품질의 다양한 신규 분자 구조를 제안하는 것이 가능하다”며, “신약 개발 초기 단계에 적용해 후보물질 및 선도물질 발굴 과정을 획기적으로 단축하고 효율성을 높일 수 있을 것으로 기대한다”고 말했다.
남호정 교수가 지도하고 배봉성 석박통합과정생과 배해리 박사과정생이 수행한 이번 연구는 한국연구재단 ‘중견연구자지원사업’ ‘원천기술개발사업’사업과 과학기술정보통신부 정보통신기술진흥센터의 지원을 받아 수행되었으며, 화학정보학 분야 저명 학술지 ‘Journal of Cheminformatics’에 2023년 9월 7일 온라인 게재됐다.